Systemy elektrochemiczne
ogólna charakterystyka
Elektrochemia - dział chemii zajmujący się badaniem procesów powstawania różnicy potencjałów i konwersji energii chemicznej na energię elektryczną (ogniwa galwaniczne), a także realizacji reakcji chemicznych w wyniku wydatkowania energii elektrycznej (elektroliza). Te dwa procesy, mające wspólny charakter, znalazły szerokie zastosowanie w nowoczesnej technologii.
Ogniwa galwaniczne wykorzystywane są jako autonomiczne i małogabarytowe źródła zasilania maszyn, urządzeń radiotechnicznych i urządzeń sterujących. Za pomocą elektrolizy uzyskuje się różne substancje, obrabiane są powierzchnie i powstają produkty o pożądanym kształcie.
Procesy elektrochemiczne nie zawsze przynoszą korzyści człowiekowi, a czasami wyrządzają wielką szkodę, powodując zwiększoną korozję i niszczenie konstrukcji metalowych. Aby umiejętnie wykorzystywać procesy elektrochemiczne i radzić sobie z niepożądanymi zjawiskami, trzeba je zbadać i umieć regulować.
Przyczyną występowania zjawisk elektrochemicznych jest przenoszenie elektronów lub zmiana stanu utlenienia atomów substancji biorących udział w procesach elektrochemicznych, czyli reakcjach redoks zachodzących w układach heterogenicznych. W reakcjach redoks elektrony są przenoszone bezpośrednio ze środka redukującego do środka utleniającego. Jeśli procesy utleniania i redukcji są przestrzennie rozdzielone, a elektrony skierowane są wzdłuż metalowego przewodnika, to takim układem będzie ogniwo galwaniczne. Przyczyną występowania i przepływu prądu elektrycznego w ogniwie galwanicznym jest różnica potencjałów.
Potencjał elektrody. Pomiar potencjałów elektrod
Jeśli weźmiesz płytkę z dowolnego metalu i zanurzysz ją w wodzie, wówczas jony warstwy powierzchniowej pod wpływem polarnych cząsteczek wody odpadną i uwodnione przejdą do cieczy. W wyniku takiego przejścia ciecz jest naładowana dodatnio, a metal jest naładowany ujemnie, ponieważ pojawia się na niej nadmiar elektronów. Akumulacja jonów metali w cieczy zaczyna spowalniać rozpuszczanie się metalu. Ustalona zostaje ruchoma równowaga
Me 0 + mH 2 O \u003d Me n + × m H 2 O + ne -
Stan równowagi zależy zarówno od aktywności metalu, jak i stężenia jego jonów w roztworze. W przypadku metali aktywnych, stojących w szeregu napięć do wodoru, oddziaływanie z cząsteczkami polarnej wody kończy się oderwaniem dodatnich jonów metali od powierzchni i przejściem jonów uwodnionych do roztworu (rys. b). Metal jest naładowany ujemnie. Procesem jest utlenianie. Wraz ze wzrostem koncentracji jonów przy powierzchni staje się możliwy proces odwrotny - redukcja jonów. Przyciąganie elektrostatyczne między kationami w roztworze a nadmiarem elektronów na powierzchni tworzy podwójną warstwę elektryczną. Prowadzi to do pojawienia się pewnej różnicy potencjałów lub skoku potencjału na granicy między metalem a cieczą. Potencjalna różnica między metalem a otaczającą go wodą nazywa się potencjał elektrody. Kiedy metal jest zanurzony w roztworze soli tego metalu, równowaga się przesuwa. Zwiększenie stężenia jonów danego metalu w roztworze ułatwia przejście jonów z roztworu do metalu. Metale, których jony mają znaczną zdolność wchodzenia do roztworu, będą w takim roztworze naładowane dodatnio, ale w mniejszym stopniu niż w czystej wodzie.
W przypadku metali nieaktywnych stężenie równowagowe jonów metali w roztworze jest bardzo niskie. Jeśli taki metal zostanie zanurzony w roztworze soli tego metalu, wówczas dodatnio naładowane jony są uwalniane na metalu szybciej niż przejście jonów z metalu do roztworu. Powierzchnia metalu otrzyma ładunek dodatni, a roztwór będzie ujemny z powodu nadmiaru anionów soli. I w tym przypadku na granicy metal-roztwór pojawia się podwójna warstwa elektryczna, stąd pewna różnica potencjałów (rys. c). W rozważanym przypadku potencjał elektrody jest dodatni.
Ryż. Proces przejścia jonu z metalu do roztworu:
równowaga; b - rozpuszczanie; c - opady
Potencjał każdej elektrody zależy od rodzaju metalu, stężenia jego jonów w roztworze oraz temperatury. Jeśli metal zostanie obniżony do roztworu jego soli zawierającego jeden mol jonu metalu na 1 dm 3 (którego aktywność wynosi 1), wówczas potencjał elektrody będzie wartością stałą w temperaturze 25 ° C i ciśnieniu 1 atm. Ten potencjał nazywa się standardowy potencjał elektrody (Eo).
Jony metali z ładunkiem dodatnim, wnikając w roztwór i poruszając się w polu potencjał interfejsu metal-roztwór, zużywają energię. Energia ta jest kompensowana pracą ekspansji izotermicznej od wyższego stężenia jonów na powierzchni do niższego stężenia w roztworze. Jony dodatnie gromadzą się w warstwie powierzchniowej do stężenia Z o, a następnie przejść do roztworu, gdzie stężenie wolnych jonów Z. Praca pola elektrycznego EnF jest równa pracy izotermicznej rozszerzania RTln(co/c). Zrównując oba przejawy pracy, możemy wyprowadzić wartość potencjału
En F \u003d RTln (c o / s), -E \u003d RTln (c / c o) / nF,
gdzie E jest potencjałem metalu, V; R jest uniwersalną stałą gazową, J/mol K; T – temperatura, K; n jest ładunkiem jonowym; F to liczba Faradaya; c jest stężeniem wolnych jonów;
с о to stężenie jonów w warstwie powierzchniowej.
Nie jest możliwe bezpośrednie zmierzenie wielkości potencjału, ponieważ nie można eksperymentalnie określić za pomocą o. Empirycznie wartości potencjałów elektrod określa się w stosunku do wartości innej elektrody, której potencjał jest warunkowo przyjmowany jako równy zero. Ta standardowa lub referencyjna elektroda jest normalna elektroda wodorowa (N.H.E.) . Urządzenie elektrody wodorowej pokazano na rysunku. Składa się z platynowej płyty pokrytej elektrolitycznie osadzoną platyną. Elektrodę zanurza się w 1M roztworze kwasu siarkowego (aktywność jonów wodorowych wynosi 1 mol/dm3) i przemywa strumieniem gazowego wodoru pod ciśnieniem 101 kPa i T = 298 K. Po nasyceniu platyny wodór, równowaga ustala się na powierzchni metalu, cały proces wyraża się równaniem
2H + + 2e ↔ H 2 .
Jeśli płytkę metalu zanurzoną w 1M roztworze soli tego metalu połączymy zewnętrznym przewodnikiem ze standardową elektrodą wodorową, a roztwory połączymy kluczem elektrolitycznym, to otrzymamy ogniwo galwaniczne (ryc. 32). Siła elektromotoryczna tego ogniwa galwanicznego będzie wartością standardowy potencjał elektrody danego metalu (E o ).

Schemat pomiaru standardowego potencjału elektrody
w stosunku do elektrody wodorowej
Biorąc cynk w 1 M roztworze siarczanu cynku jako elektrodę i podłączając go do elektrody wodorowej, otrzymujemy ogniwo galwaniczne, którego obwód piszemy następująco
(-) Zn/Zn2+ //2H+/H2, Pt(+).
Na schemacie jedna linia wskazuje granicę między elektrodą a roztworem, dwie linie - granicę między roztworami. Anoda jest napisana po lewej stronie, katoda po prawej. W takim elemencie zachodzi reakcja Zn o + 2H + \u003d Zn 2+ + H 2, a elektrony przechodzą przez obwód zewnętrzny od cynku do elektrody wodorowej. Standardowy potencjał elektrody cynkowej (-0,76 V).
Biorąc jako elektrodę płytkę miedzianą, we wskazanych warunkach, w połączeniu ze standardową elektrodą wodorową otrzymujemy ogniwo galwaniczne
(-) Pt, H2/2H+//Cu2+/Cu (+).
W tym przypadku reakcja przebiega: Cu 2+ + H 2 = Cu o + 2H +. Elektrony przemieszczają się wzdłuż obwodu zewnętrznego od elektrody wodorowej do elektrody miedzianej. Standardowy potencjał elektrody miedzianej (+0,34 V).
Szereg standardowych potencjałów elektrod (napięcia). Równanie Nernsta
Układając metale w porządku rosnącym ich standardowych potencjałów elektrod, otrzymuje się szereg napięć Nikołaja Nikołajewicza Beketowa (1827-1911) lub szereg standardowych potencjałów elektrod. Wartości liczbowe standardowych potencjałów elektrod dla wielu technicznie ważnych metali podano w tabeli.
Szereg metali naprężeniowych

Szereg naprężeń charakteryzuje niektóre właściwości metali:
1. Im mniejsza wartość potencjału elektrodowego metalu, im bardziej jest on aktywny chemicznie, tym łatwiej się utlenia i tym trudniej jest odzyskać z jego jonów. Metale aktywne w przyrodzie występują tylko w postaci związków Na, K, ..., występują w naturze zarówno w postaci związków, jak iw stanie wolnym Cu, Ag, Hg; Au, Pt - tylko w stanie wolnym;
2. Metale, które mają bardziej ujemny potencjał elektrody niż magnez, wypierają wodór z wody;
3. Metale stojące w szeregu napięć do wodoru wypierają wodór z roztworów rozcieńczonych kwasów (których aniony nie wykazują właściwości utleniających);
4. Każdy metal z serii, który nie rozkłada wody, wypiera z roztworów ich soli metale, które mają bardziej dodatnie wartości potencjałów elektrod;
5. Im bardziej metale różnią się wartościami potencjałów elektrod, tym większa wartość emf. będzie miał zbudowane z nich ogniwo galwaniczne.
Zależność potencjału elektrody (E) od charakteru metalu, aktywności jego jonów w roztworze i temperatury wyraża równanie Nernsta
E Me \u003d E o Me + RTln (a Me n +) / nF,
gdzie E o Me jest standardowym potencjałem elektrody metalu, a Me n + jest aktywnością jonów metalu w roztworze. W standardowej temperaturze 25 o C, dla rozcieńczonych roztworów, zastępując aktywność (a) stężeniem (c), logarytm naturalny do ułamka dziesiętnego i podstawiając wartości R, T i F, otrzymujemy
E Me \u003d E o Me + (0,059 / n) lgc.
Np. dla elektrody cynkowej umieszczonej w roztworze jej soli stężenie uwodnionych jonów Zn 2+ × mH 2 O w skrócie Zn 2+ , wtedy
E Zn \u003d E o Zn + (0,059 / n) lg [ Zn 2+ ].
Jeśli \u003d 1 mol / dm 3, to E Zn \u003d E o Zn.
Ogniwa galwaniczne, ich siła elektromotoryczna
Dwa metale zanurzone w roztworach ich soli, połączone przewodem, tworzą ogniwo galwaniczne. Pierwsze ogniwo galwaniczne zostało wynalezione przez Alexandra Volta w 1800 roku. Ogniwo składało się z płyt miedzianych i cynkowych oddzielonych tkaniną nasączoną kwasem siarkowym. Gdy duża liczba płyt jest połączona szeregowo, element Volta ma znaczną siłę elektromotoryczną (emf).
Występowanie prądu elektrycznego w ogniwie galwanicznym wynika z różnicy potencjałów elektrod pobieranych metali i towarzyszą mu przemiany chemiczne zachodzące na elektrodach. Rozważ działanie ogniwa galwanicznego na przykładzie ogniwa miedziano-cynkowego (J. Daniel - B.S. Jacobi).

Schemat miedziano-cynkowego ogniwa galwanicznego Daniela-Jacobiego
Na elektrodzie cynkowej zanurzonej w roztworze siarczanu cynku (c = 1 mol / dm 3) cynk jest utleniany (cynk rozpuszcza się) Zn o - 2e = Zn 2+. Elektrony wchodzą do obwodu zewnętrznego. Zn jest źródłem elektronów. Za źródło elektronów uważa się elektrodę ujemną – anodę. Na elektrodzie miedzianej zanurzonej w roztworze siarczanu miedzi (c \u003d 1 mol / dm 3) jony metali ulegają redukcji. Atomy miedzi osadzają się na elektrodzie Cu 2+ + 2e = Cu o. Elektroda miedziana jest dodatnia. To jest katoda. Jednocześnie część jonów SO 4 2- przechodzi przez mostek solny do naczynia z roztworem ZnSO 4 . Dodając równania procesów zachodzących na anodzie i katodzie otrzymujemy równanie sumaryczne
|
Boris Semenovich Jacobi (Moritz Herman) (1801-1874) |

lub w formie molekularnej
Jest to powszechna reakcja redoks, która zachodzi na granicy metal-roztwór. Energia elektryczna ogniwa galwanicznego jest uzyskiwana w wyniku reakcji chemicznej. Rozważane ogniwo galwaniczne można zapisać w postaci krótkiego obwodu elektrochemicznego
(-) Zn/Zn 2+ //Cu 2+ /Cu (+).
Niezbędnym warunkiem działania ogniwa galwanicznego jest różnica potencjałów, która nazywa się siła elektromotoryczna ogniwa galwanicznego (emf) . emf dowolnego działającego ogniwa galwanicznego, wartość jest dodatnia. Aby obliczyć emf. ogniwa galwanicznego należy odjąć wartość potencjału mniej dodatniego od wartości potencjału bardziej dodatniego. Więc e.f.s. miedziano-cynkowe ogniwo galwaniczne w standardowych warunkach (t \u003d 25 ° C, c \u003d 1 mol / dm 3, P \u003d 1 atm) jest równe różnicy między standardowymi potencjałami elektrod miedzi (katody) i cynku (anody ), to znaczy
emf = E o C u 2+ / Cu - E o Zn 2+ / Zn = +0,34 V - (-0,76 V) = +1,10 V.
W połączeniu z cynkiem jon Cu 2+ jest zredukowany.
Niezbędną do działania różnicę potencjałów elektrod można wytworzyć przy użyciu tego samego roztworu o różnych stężeniach i tych samych elektrod. Takie ogniwo galwaniczne nazywa się stężenie , i działa poprzez wyrównywanie stężeń roztworu. Przykładem jest element składający się z dwóch elektrod wodorowych.
Pt, H 2 / H 2 SO 4 (c`) // H 2 SO 4 (c``) / H 2, Pt,
gdzie c` = `; c`` = ``.
Jeśli p = 101 kPa, s`< с``, то его э.д.с. при 25 о С определяется уравнением
E = 0,059lg (s``/s`).
Z c` \u003d 1 mol-ion / dm 3 emf. pierwiastek zależy od stężenia jonów wodorowych w drugim roztworze, to znaczy E \u003d 0,059lgc `` \u003d -0,059 pH.
Oznaczanie stężenia jonów wodorowych, a w konsekwencji pH podłoża poprzez pomiar siły elektromotorycznej. odpowiednie ogniwo galwaniczne nazywa się potencjometrią.
Baterie
Baterie nazywane są ogniwami galwanicznymi wielokrotnego i odwracalnego działania. Są w stanie zamienić nagromadzoną energię chemiczną na energię elektryczną podczas rozładowywania, a energię elektryczną na energię chemiczną, tworząc rezerwę podczas procesu ładowania. Od emf baterie są małe, podczas pracy są zwykle połączone z bateriami.
Akumulator ołowiowy . Akumulator ołowiowy składa się z dwóch perforowanych płytek ołowiowych, z których jedna (ujemna) po naładowaniu zawiera wypełniacz - gąbka aktywny ołów, a druga (dodatnia) - dwutlenek ołowiu. Obie płytki zanurza się w 25-30% roztworze kwasu siarkowego (ryc. 35). Schemat baterii
(-) Pb/ p -p H 2 SO 4 / PbO 2 /Pb (+) .
Przed naładowaniem w pory elektrod ołowiowych nasmarowuje się pastę zawierającą oprócz spoiwa organicznego tlenek ołowiu PbO. W wyniku oddziaływania tlenku ołowiu z kwasem siarkowym w porach płyt elektrody powstaje siarczan ołowiu.
PbO + H2SO4 \u003d PbSO4 + H2O .
Baterie są ładowane poprzez przepływ prądu elektrycznego

Proces rozładowywania

W sumie procesy zachodzące podczas ładowania i rozładowywania akumulatora można przedstawić w następujący sposób:
Gdy akumulator jest naładowany, gęstość elektrolitu (kwasu siarkowego) wzrasta, a po rozładowaniu maleje. Gęstość elektrolitu służy do oceny stopnia rozładowania akumulatora. emf akumulator ołowiowy 2,1 V.
Zalety akumulator ołowiowy - duża pojemność elektryczna, stabilność pracy, duża liczba cykle (rozładowanie-rozładowanie). Wady- duża masa, a w konsekwencji mała pojemność właściwa, wydzielanie wodoru podczas ładowania, brak szczelności w obecności stężonego roztworu kwasu siarkowego. Pod tym względem baterie alkaliczne są lepsze.
Baterie alkaliczne. Należą do nich baterie kadmowo-niklowe i żelazowo-niklowe T. Edisona.

Schematy akumulatorów Edisona i akumulatorów ołowiowych
|
Tomasz Edison(1847-1931) |
Są do siebie podobne. Różnica polega na materiale płyt elektrod ujemnych. W pierwszym przypadku są to kadm, w drugim żelazo. Elektrolit to roztwór KOH ω = 20% . Największe znaczenie praktyczne mają akumulatory kadmowo-niklowe. Schemat baterii kadmowo-niklowej
(-) roztwór Cd/KOH/Ni2O3/Ni(+).
Działanie baterii kadmowo-niklowej opiera się na reakcji redoks z udziałem Ni 3+
emf naładowany akumulator niklowo-kadmowy wynosi 1,4 V.
W tabeli przedstawiono charakterystykę akumulatora Edison i akumulatora ołowiowego.
W ogniwie elektrochemicznym (ogniwo galwaniczne) elektrony pozostałe po utworzeniu jonów są usuwane przez metalowy drut i rekombinowane z jonami innego rodzaju. Oznacza to, że ładunek w obwodzie zewnętrznym jest przenoszony przez elektrony, a wewnątrz ogniwa przez elektrolit, w którym zanurzone są elektrody metalowe, przez jony. W ten sposób uzyskuje się zamknięty obwód elektryczny.
Różnica potencjałów mierzona w ogniwie elektrochemicznym, o ze względu na różnicę w zdolności każdego z metali do oddawania elektronów. Każda elektroda ma swój własny potencjał, każdy układ elektroda-elektrolit jest półogniwem, a dowolne dwa półogniwa tworzą ogniwo elektrochemiczne. Potencjał jednej elektrody nazywany jest potencjałem półogniwa, określa on zdolność elektrody do oddawania elektronów. Oczywiście potencjał każdego półelementu nie zależy od obecności innego półelementu i jego potencjału. Potencjał półogniwowy jest określony przez stężenie jonów w elektrolicie i temperaturę.
Jako półelement „zerowy” wybrano wodór; zakłada się, że nie wykonuje się dla niego żadnej pracy, gdy elektron jest dodawany lub usuwany w celu utworzenia jonu. Wartość „zero” potencjału jest niezbędna do zrozumienia względnej zdolności każdego z dwóch półelementów komórki do dawania i odbierania elektronów.
Potencjały półogniw mierzone względem elektrody wodorowej nazywane są skalą wodoru. Jeżeli termodynamiczna skłonność do oddawania elektronów w jednej połowie ogniwa elektrochemicznego jest wyższa niż w drugiej, to potencjał pierwszej połowy ogniwa jest wyższy niż potencjał drugiej. Pod działaniem różnicy potencjałów nastąpi przepływ elektronów. Kiedy dwa metale są połączone, możliwe jest określenie różnicy potencjałów między nimi i kierunku przepływu elektronów.
Metal elektrododatni ma wyższą zdolność przyjmowania elektronów, więc będzie katodowy lub szlachetny. Z drugiej strony istnieją metale elektroujemne, które są zdolne do spontanicznego oddawania elektronów. Te metale są reaktywne, a zatem anodowe:
- → 0 → +
Al Mn Zn Fe Sn Pb H 2 Cu Ag Au
Na przykład Cu łatwiej oddawać elektrony Ag, ale gorzej niż Fe . W obecności elektrody miedzianej, srebrne nony zaczną łączyć się z elektronami, prowadząc do powstania jonów miedzi i wytrącenia metalicznego srebra:
2 Ag + + Cu → Cu 2+ + 2 Ag
Jednak ta sama miedź jest mniej reaktywna niż żelazo. Kiedy metaliczne żelazo wejdzie w kontakt z miedzią, wytrąci się, a żelazo przejdzie do roztworu:
Fe + Cu 2+ → Fe 2+ + Cu.
Można powiedzieć, że miedź jest metalem katodowym w stosunku do żelaza i metalem anodowym w stosunku do srebra.
Za standardowy potencjał elektrody uważa się potencjał w pełni wyżarzonego półogniwa z czystego metalu jako elektrody w kontakcie z jonami w temperaturze 25°C. W tych pomiarach elektroda wodorowa działa jako elektroda odniesienia. W przypadku metalu dwuwartościowego reakcję zachodzącą w odpowiednim ogniwie elektrochemicznym można zapisać:
M + 2H +→ M 2+ + H 2 .
Jeśli metale są uporządkowane w porządku malejącym ich standardowych potencjałów elektrod, otrzymuje się tak zwany elektrochemiczny szereg napięć metali (tabela 1).
Tabela 1. Elektrochemiczne szeregi napięć metali
|
Równowaga jonów metali (pojedyncza aktywność) |
Potencjał elektrody względem elektrody wodorowej przy 25°С, V (potencjał redukcyjny) |
|
|
szlachetny lub katodowy |
Au-Au 3+ |
1,498 |
|
Pt-Pt 2+ |
||
|
Pd-Pd 2+ |
0,987 |
|
|
Ag-Ag+ |
0,799 |
|
|
Hg-Hg 2+ |
0,788 |
|
|
Cu-Cu 2+ |
0,337 |
|
|
H2-H+ |
||
|
Pb-Pb 2+ |
0,126 |
|
|
Sn-Sn 2+ |
0,140 |
|
|
Ni-Ni 2+ |
0,236 |
|
|
CoCo 2+ |
0,250 |
|
|
Cd-Cd 2+ |
0,403 |
|
|
Fe-Fe 2+ |
0,444 |
|
|
Cr-Cr 2+ |
0,744 |
|
|
Zn-Zn 2+ |
0,763 |
|
|
Aktywny |
Al-Al2+ |
1,662 |
|
Mg-Mg2+ |
2,363 |
|
|
Na-Na + |
2,714 |
|
|
K-K+ |
2,925 |
Na przykład w ogniwie galwanicznym miedziano-cynkowym następuje przepływ elektronów z cynku do miedzi. Elektroda miedziana jest biegunem dodatnim w tym obwodzie, a elektroda cynkowa jest biegunem ujemnym. Bardziej reaktywny cynk traci elektrony:
Zn → Zn 2+ + 2e - ; E°=+0,763 V.
Miedź jest mniej reaktywna i przyjmuje elektrony z cynku:
Cu 2+ + 2e - → Cu; E°=+0,337 V.
Napięcie na metalowym przewodzie łączącym elektrody będzie wynosić:
0,763V + 0,337V = 1,1V.
Tabela 2. Potencjały stacjonarne niektórych metali i stopów w wodzie morskiej w odniesieniu do normalnej elektrody wodorowej (GOST 9.005-72).
|
Metal |
Potencjał stacjonarny, W |
Metal |
Potencjał stacjonarny, W |
|
Magnez |
1,45 |
Nikiel (aktywny co stoi) |
0,12 |
|
Stop magnezu (6% A ja , 3 % Zn, 0,5 % Mn) |
1,20 |
Stopy miedzi LMtsZh-55 3-1 |
0,12 |
|
Cynk |
0,80 |
Mosiądz (30 % Zn) |
0,11 |
|
Stop aluminium (10% Mn) |
0,74 |
Brązowy (5-10 % Glin) |
0,10 |
|
Stop aluminium (10% Zn) |
0,70 |
Czerwony mosiądz (5-10 % Zn) |
0,08 |
|
Stop aluminium K48-1 |
0,660 |
Miedź |
0,08 |
|
Stop aluminium B48-4 |
0,650 |
miedzionikiel (30% Ni) |
0,02 |
|
Stop aluminium AMg5 |
0,550 |
Brązowa „Nowa” |
0,01 |
|
Stop aluminium AMg61 |
0,540 |
Brąz Fr. AJN 9-4-4 |
0,02 |
|
Aluminium |
0,53 |
Stal nierdzewna X13 (stan pasywny) |
0,03 |
|
Kadm |
0,52 |
Nikiel (stan pasywny) |
0,05 |
|
Duraluminium i stop aluminium AMg6 |
0,50 |
Stal nierdzewna X17 (stan pasywny) |
0,10 |
|
Żelazo |
0,50 |
Techniczny tytan |
0,10 |
|
Stal 45G17Yu3 |
0,47 |
Srebro |
0,12 |
|
Stal St4S |
0,46 |
Stal nierdzewna 1X14ND |
0,12 |
|
Stal SHL4 |
0,45 |
Jodek tytanu |
0,15 |
|
Stal typu AK i stal węglowa |
0,40 |
Stal nierdzewna Kh18N9 (stan pasywny) i OH17N7Yu |
0,17 |
|
Żeliwo szare |
0,36 |
metal monel |
0,17 |
|
Stal nierdzewna X13 i X17 (stan aktywny) |
0,32 |
Stal nierdzewna Х18Н12М3 (stan pasywny) |
0,20 |
|
Żeliwo niklowo-miedziowe (12-15% Ni, 5-7% Si) |
0,30 |
Stal nierdzewna Х18Н10Т |
0,25 |
|
Prowadzić |
0,30 |
Platyna |
0,40 |
|
Cyna |
0,25 |
Notatka . Podane wartości liczbowe potencjałów i kolejność metali w szeregu mogą różnić się w różnym stopniu w zależności od czystości metali, składu wody morskiej, stopnia napowietrzenia i stanu powierzchni metale.
Do metali zaliczamy pierwiastki s z grup 1 i 2, wszystkie pierwiastki d i f oraz szereg pierwiastków p z głównych podgrup: 3 (oprócz boru), 4 (Ge, Sn, Pb), 5 (Sb, Bi) i Ro. Najbardziej typowe elementy metalowe znajdują się na początku epok. Wcześniej mówiliśmy o tym, że w metalach zachodzi silnie zdelokalizowane wiązanie. Wynika to z faktu, że ze względu na efekt ekranowania elektrony walencyjne w atomach metali są słabiej przyciągane do jądra, a pierwsze energie jonizacji dla nich są stosunkowo niewielkie. W normalnej dla nas temperaturze (około 300 K), która jest dość daleka od zera absolutnego, energia ruchu termicznego jest wystarczająca do swobodnego przemieszczania się elektronów w metalu.
Ponieważ wiązanie w metalach jest silnie zdelokalizowane i rozciąga się na cały kryształ, metale mają wysoką plastyczność, przewodnictwo elektryczne i cieplne. Srebro i miedź mają najwyższą przewodność elektryczną i cieplną, a rtęć najniższą. Ten ostatni jest również najbardziej topliwym metalem (-38,9 C). najbardziej ogniotrwałym metalem jest wolfram (3390 C). Tak dużą różnicę temperatur topnienia i wrzenia tłumaczy się obecnością w metalach, oprócz wiązania metalicznego, pewnej proporcji wiązań kowalencyjnych, zwłaszcza dla pierwiastków przejściowych, które mają dużą liczbę elektronów walencyjnych.
Rozważ elektroniczne konfiguracje rtęci i wolframu.
Hg – 5d 10 6s 2 ; W - 5d 4 6s 2 . Oddziaływanie międzycząsteczkowe między atomami rtęci jest bardzo małe, tak małe, że na ogół przy dużej gęstości, ze względu na grawitację atomów, jest to najbardziej topliwy metal. Ponieważ wszystkie podpoziomy w atomie rtęci są wypełnione, tworzenie wiązań kowalencyjnych jest generalnie niemożliwe, a wiązanie metaliczne jest raczej słabe, słabsze niż w metalach alkalicznych, które są na ogół najbardziej topliwe spośród wszystkich metali. Wręcz przeciwnie, w atomie W możliwe jest utworzenie czterech wiązań walencyjnych na raz. Ponadto wiązanie metaliczne jest najsilniejsze spośród wszystkich pierwiastków 5d, a same atomy są cięższe niż ich elektronowe odpowiedniki: Mo i Cr. Połączenie tych czynników prowadzi do najwyższej ogniotrwałości wolframu.
Konfiguracja elektronowa osmu (5d 6 6s 2) jest taka, że brakuje mu 4 elektronów przed ukończeniem podpoziomu 5d, dlatego jest najbardziej zdolny do przyciągania elektronów z sąsiednich atomów, co powoduje skrócenie wiązania metal-metal. Dlatego osm ma najwyższą gęstość (22,4 g / cm 3).
W czystej postaci metale są stosunkowo rzadkie. Zasadniczo są to metale chemicznie obojętne (złoto, a także metale z grupy platynowców - platyna, rod, iryd, osm itp.). Srebro, miedź, rtęć, cyna mogą występować zarówno w stanie natywnym, jak i w postaci związków. Pozostałe metale występują w postaci związków zwanych rudami.
Metale otrzymuje się z ich związków, redukując je z tlenków. Jako czynniki redukujące stosuje się C, CO, metale aktywne, wodór, metan. Jeśli siarczek metalu (ZnS, FeS 2) działa jak ruda, to najpierw jest przekształcany w tlenek. Redukcja metali z ich związków przez inne metale nazywa się metalotermią. Niektóre metale są ekstrahowane z roztworów ich soli za pomocą elektrolizy, takich jak aluminium lub sód. Wszystkie metody otrzymywania metali z ich związków opierają się na procesach redoks.
Proces przeniesienia elektronu w połówkowej reakcji redoks można przedstawić następującym ogólnym równaniem:
Procesowi przenoszenia elektronów odpowiada zmiana energii Gibbsa, równa ∆G = –nFE, gdzie F (stała Faradaya odpowiada ilości energii elektrycznej potrzebnej do redukcji lub utlenienia jednego mola substancji) = 96500 C/mol , n to liczba elektronów, E to potencjał elektrody, V to różnica napięć między środkiem utleniającym a środkiem redukującym. Z drugiej strony ∆G = –RTlnK = –nFE; RTlnK = nFE. Stąd E = RTlnK/nF. Ponieważ K = / i 2,3lnK = lgK, zależność potencjału elektrody od stężeń substancji - uczestników procesu elektrodowego - i temperatury wyraża się następującym równaniem:
E = E 0 + lg/ jest równaniem Nernsta.
W temperaturze standardowej (298 K) równanie staje się:
E \u003d E 0 + 0,059 g /
W liczniku zawsze podaje się stężenie środka utleniającego, a potencjał połówkowej reakcji redukcji: Ox + ne = Red.
Przy równowagowych stężeniach środka utleniającego i środka redukującego równych jeden, E \u003d E 0 jest standardowym potencjałem elektrody: jest to potencjał danego procesu elektrodowego przy stężeniach jednostkowych wszystkich substancji. Ponieważ niemożliwe jest określenie wartości bezwzględnej potencjałów elektrod wzorcowych, za punkt odniesienia przyjmuje się potencjał półreakcyjny: 2H + + 2e = H 2 . Potencjał tego procesu elektrodowego przyjmuje się jako równy 0 przy jednostkowych stężeniach kationu wodoru. Elektroda wodorowa składa się z płytki platynowej, którą zanurza się w roztworze kwasu siarkowego o [H+] = 1 mol/l i przemywa strumieniem H2 pod ciśnieniem 101325 Pa w temperaturze 298 K.
Potencjał elektrody nazywa się EMF ogniwa galwanicznego, które składa się z badanej elektrody i standardowej elektrody wodorowej. Układając metale w kolejności rosnącej wielkości ich potencjałów elektrodowych, otrzymujemy szereg standardowych potencjałów elektrodowych metali. Charakteryzuje właściwości chemiczne metali. Każdy metal w serii wypiera wszystkie kolejne metale z roztworu ich soli. Metale w rzędzie na lewo od wodoru wypierają go z kwaśnych roztworów.
Potencjał dowolnej reakcji redoks można obliczyć z wartości potencjałów połówkowych reakcji.
Rozważ prosty przykład: Zn + 2HCl = ZnCl 2 + H 2 . W tym procesie występują dwie reakcje połowiczne:
Zn 2+ + 2e \u003d Zn 0 E 0 (Zn 2+ / Zn) \u003d -0,76 B
2H + + 2e \u003d H 2 0 E 0 (2H + /H 2) \u003d 0,00 B
Ponieważ potencjał drugiej połówkowej reakcji jest wyższy niż pierwszej, druga połówkowa reakcja będzie przebiegać od lewej do prawej, to znaczy w kierunku tworzenia cząsteczek wodoru. Pierwsza reakcja połowiczna przebiegać będzie od prawej do lewej, czyli w kierunku powstawania kationów cynku.
Rozważając produkcję metali, powiedzieliśmy, że wiele metali jest redukowanych z ich tlenków przez inne, bardziej aktywne metale. Na przykład magnez może redukować miedź z tlenku miedzi(II). Porównajmy dwie reakcje połowiczne:
Cu 2+ + 2e = Cu E 0 = +0,34 V
Mg 2+ + 2e \u003d Mg E 0 \u003d -2,36 V
Potencjał pierwszej reakcji połowicznej jest wyższy niż drugiej i to ona będzie postępować od lewej do prawej, a druga - od prawej do lewej.
Zatem, aby określić kierunek reakcji redoks, należy zapisać dwie połówkowe reakcje z formy utlenionej do zredukowanej i porównać ich potencjały. Reakcja, której potencjał jest wyższy, będzie przebiegać od lewej do prawej, a ta o niższym potencjale będzie postępować od prawej do lewej.
Prawie wszystkie reakcje z metalami są procesami redoks, a aby określić ich kierunek, należy przede wszystkim wziąć pod uwagę potencjały każdej z reakcji połówkowych w procesie redoks. Ale poza tym są wyjątki. Na przykład ołów jest nierozpuszczalny w kwasie siarkowym, mimo że potencjał pary Pb 2+ /Pb wynosi –0,15 V. Faktem jest, że siarczan ołowiu jest nierozpuszczalny i jego powstawanie zapobiega dalszemu utlenianiu ołowiu.
Wykład 15
Elektroliza.
Roztwory i stopione elektrolity zawierają przeciwnie naładowane jony (kationy i aniony), które są w ciągłym ruchu. Jeśli elektrody obojętne (grafitowe) zostaną zanurzone w takiej cieczy, na przykład w stopionym chlorku sodu (topi się w 801 0 C) i przepłynie stały prąd elektryczny, to jony pod działaniem zewnętrznego pola elektrycznego przesuną się do elektrody, kationy - do katody i aniony - do anody. Kationy sodu po osiągnięciu katody przyjmują z niej elektrony i są redukowane do metalicznego sodu:
Jony chlorkowe są utleniane na anodzie:
2Cl - - 2e \u003d Cl 2 0
W rezultacie metaliczny sód jest uwalniany na katodzie, a cząsteczkowy chlor jest uwalniany na anodzie. Ogólne równanie elektrolizy stopionego chlorku sodu jest następujące.
K: Na + + e \u003d Na 0 2
ALE: 2Cl - - 2e \u003d Cl 2 0 1
2Na + + 2Cl - elektroliza ® 2Na 0 + Cl 2 0
2NaCl \u003d 2Na + Cl 2
Ta reakcja jest reakcją redoks: proces utleniania zachodzi na anodzie, a proces redukcji zachodzi na katodzie.
Proces redoks, który zachodzi na elektrodach, gdy prąd elektryczny przepływa przez roztwór stopu lub elektrolitu, nazywa się elektrolizą.
Istotą elektrolizy jest realizacja reakcji chemicznych pod wpływem energii elektrycznej. W tym przypadku katoda oddaje elektrony kationom, a anoda przyjmuje elektrony z anionów. Działanie stałego prądu elektrycznego jest znacznie silniejsze niż działanie chemicznych środków redukujących i utleniających. To właśnie przez elektrolizę po raz pierwszy udało się uzyskać gazowy fluor.
Elektrolizę prowadzono w roztworze fluorku potasu w kwasie fluorowodorowym. W tym przypadku na anodzie uwalniany jest fluor, a na katodzie wodór. Elektrolizę przeprowadza się w kąpieli elektrolitycznej.
Konieczne jest rozróżnienie pomiędzy elektrolizą stopionych elektrolitów a ich roztworami. W tym ostatnim przypadku w procesach mogą uczestniczyć cząsteczki wody. Na przykład podczas elektrolizy wodnego roztworu chlorku sodu na elektrodach obojętnych (grafitowych) cząsteczki wody są redukowane na katodzie zamiast kationów sodu.
2H 2 O + 2e \u003d H 2 + 2OH -
a jony chlorkowe utleniają się na anodzie:
2Cl - - 2e \u003d Cl 2 0
W rezultacie na katodzie uwalniany jest wodór, na anodzie uwalniany jest chlor, aw roztworze gromadzą się cząsteczki wodorotlenku sodu. Ogólne równanie elektrolizy wodnego roztworu chlorku sodu to:
K: 2H 2 O + 2e \u003d H 2 + 2OH -
ALE: 2Cl - - 2e \u003d Cl 2 0
2H2O + 2Cl - \u003d H2 + Cl2 + 2OH -
Nawiasem mówiąc, w ten sposób w przemyśle otrzymuje się wodorotlenki wszystkich metali alkalicznych i niektórych metali ziem alkalicznych, a także aluminium.
Jaka jest różnica między elektrolizą stopów a wodnymi roztworami elektrolitów? Procesy redukcji na katodzie wodnych roztworów elektrolitów zależą od wartości standardowych potencjałów elektrod metali, a mianowicie działają najczęściej jako kationy redukujące się na katodzie. Są tu trzy opcje:
1. Kationy metali, które mają standardowy potencjał elektrody wyższy niż wodoru, czyli większy niż zero, są całkowicie redukowane na katodzie podczas elektrolizy (miedź, srebro, złoto i inne).
2. Kationy metali o bardzo niskiej wartości potencjału elektrody wzorcowej (od litu do aluminium włącznie) nie są redukowane na katodzie, ale cząsteczki wody są redukowane.
3. Kationy metali, w których wartość potencjału elektrody standardowej jest mniejsza niż wodoru, ale większa niż aluminium, ulegają redukcji na katodzie podczas elektrolizy wraz z cząsteczkami wody.
Jeżeli kilka kationów metali znajduje się jednocześnie w roztworze wodnym, to podczas elektrolizy ich izolacja na katodzie przebiega w celu zmniejszenia wartości algebraicznej potencjału elektrody wzorcowej odpowiedniego metalu. Na przykład przy analizie brązu typu BrAZh lub BrAZhMts (miedź, aluminium, żelazo i mangan) można, wybierając określoną wartość natężenia prądu, oddzielić miedź na elektrodę obojętną (np. platynową), pociągnąć wyjmij elektrodę, zważ ją i określ zawartość miedzi. Następnie oddziel aluminium, określ jego zawartość. W ten sposób dobrze jest oddzielić metale o dodatniej wartości potencjału elektrody wzorcowej.
Wszystkie elektrody są podzielone na nierozpuszczalne (obojętne) - węgiel, grafit, platyna, iryd. Rozpuszczalny - miedź, srebro, cynk, kadm, nikiel i inne. Pojęcie rozpuszczalnej elektrody jest ważne dla anody, ponieważ to ona jest w stanie rozpuścić się podczas elektrolizy. Na nierozpuszczalnej anodzie podczas elektrolizy aniony lub cząsteczki wody ulegają utlenieniu. W tym przypadku aniony kwasów beztlenowych dość łatwo ulegają utlenieniu. Jeżeli w roztworze znajdują się aniony kwasów zawierających tlen, wówczas cząsteczki wody utleniają się na anodzie z uwolnieniem tlenu zgodnie z reakcją:
2H 2 O - 4e \u003d O 2 + 4H +
Rozpuszczalna anoda utlenia się podczas elektrolizy, oddając elektrony do zewnętrznego obwodu elektrycznego i przechodząc do roztworu:
A: Ja Û Ja n+ + ne –
Rozważmy przykłady elektrolizy stopów i roztworów elektrolitów.
Ryż. 128. Urządzenie do pomiaru normalnego potencjału metalu
Istnieje kilka teorii wyjaśniających występowanie prądu w ogniwach galwanicznych. Najprostszy z nich został przedstawiony przez Nernsta (1888), a później szczegółowo rozwinięty przez akademika L. V. Pisarzhevsky'ego na podstawie idei budowy metali z dodatnio naładowanych jonów i wolnych elektronów.
Lew Władimirowicz Pisarzewski urodził się w 1874 roku. Kiszyniów. Po ukończeniu wydziału przyrodniczego Uniwersytetu Noworosyjskiego (Odessa) Pisarzewski pozostał z nim, aby przygotować się do tytułu wielkiego profesora. W 1902 obronił pracę magisterską, aw 1913 został wybrany profesorem Jekaterynosławskiego Instytutu Górniczego (Dniepropietrowsk). Od 1930 r. Pisarzewski był pełnoprawnym członkiem Akademii Nauk ZSRR.
Wybitny naukowiec i genialny nauczyciel Pisarzhevsky śmiało wykorzystywał osiągnięcia fizyki do badania i wyjaśniania procesów chemicznych. Jego najważniejsze prace poświęcone są badaniu nadtlenków i nadkwasów, rozwojowi teorii roztworów, zastosowaniu teorii elektronów w chemii oraz rozwojowi teorii pojawiania się prądu w ogniwach galwanicznych.
Występowanie prądu w ogniwie galwanicznym następuje w następujący sposób. Jeśli jakikolwiek metal zostanie zanurzony w wodzie, jego jony zaczynają rozpuszczać się pod wpływem przyciągania od strony polarnych cząsteczek wody. W rezultacie metalnadmiar elektronów pozostaje, jest naładowany ujemnie, a roztwór jest naładowany dodatnio. Jednak liczba jonów, które metal wysyła do roztworu, jak pokazuje doświadczenie, jest bardzo mała. Ujemny ładunek, który powstaje na metalu, gdy jony opuszczają metal, zaczyna przyciągać z powrotem jony, które opuściły metal, tak że wkrótce wchodzi w stan równowagi, w którym tyle jonów opuszcza metal w jednostce czasu, gdy wracają do to:
metal (jony metali)
(w rozwiązaniu)
Jony, które przeszły do roztworu, nie są rozłożone równomiernie w masie roztworu, ale z powodu przyciągania do ujemnie naładowanego metalu znajdują się w pobliżu jego powierzchni, tworząc tak zwaną podwójną warstwę elektryczną (ryc. 127). W rezultacie powstaje pewna potencjalna różnica między metalem a roztworem.

Lew Władimirowicz Pisarzewski (1874-1938)
Załóżmy teraz, że do wody, w której zanurzony jest metal, dodaliśmy pewną ilość soli tego samego metalu. Ze względu na wzrost stężenia jonów metali w roztworze, równowaga między nimi a metalem zostanie zaburzona i część jonów wróci do metalu. Dlatego w roztworze swojej soli
metal powinien wysyłać mniej jonów niż w czystej wodzie, a im mniej, tym większe stężenie jonów w roztworze. Jeśli stężenie soli jest wystarczająco wysokie, jony mogą w ogóle nie przejść z metalu do roztworu, tak że ani metal, ani roztwór nie zostaną naładowane.
Wreszcie, jeśli stężenie jonów metali w roztworze jest wystarczająco wysokie, a aktywność metalu jest stosunkowo niska, metal nie tylko nie wysyła jonów do roztworu, ale wręcz przeciwnie, część jonów przechodzi z roztwór do metalu. W tym przypadku również powstaje różnica potencjałów między metalem a roztworem, ale teraz roztwór jest naładowany ujemnie z powodu nadmiaru ujemnych jonów soli, a metal jest naładowany dodatnio. W praktyce sytuacja jest taka, że niektóre (bardziej aktywne) są zawsze naładowane ujemnie w roztworach swoich soli, podczas gdy inne (mniej aktywne) są naładowane dodatnio.
Należy zauważyć, że we wszystkich przypadkach, gdy metal jest zanurzony w roztworze jego soli, ilość jonów przechodzących do roztworu lub uciekających z roztworu jest tak mała, że nie można ich wykryć chemicznie. Jednak ich ładunek jest na tyle duży, że tworzy mierzalną różnicę potencjałów.
Powyższa teoria bardzo prosto wyjaśnia mechanizm działania ogniw galwanicznych. Rozważmy na przykład element miedziano-cynkowy. W tym elemencie na płytce cynkowej zanurzonej w roztworze ZnSO4 powstaje pewien ładunek ujemny, a na miedzi zanurzonej w roztworze CuSO4 powstaje ładunek dodatni. Jeśli nie są one połączone ze sobą przewodem, pojawienie się tych ładunków, jak widzieliśmy powyżej, powinno natychmiast zatrzymać zarówno dalsze przejście jonów cynku do roztworu, jak i uwalnianie jonów miedzi z roztworu. Ale jeśli połączysz obie płytki drutem, to elektrony gromadzące się na cynku będą cały czas płynąć do miedzianej płytki, gdzie ich brakuje. W ten sposób możliwe staje się przesyłanie do roztworu coraz większej ilości jonów Zn, podczas gdy na płycie miedzianej jony Cu są rozładowywane i uwalniane w postaci metalicznej miedzi. Proces ten trwa aż do całkowitego lub całkowitego rozpuszczenia lub zużycia soli miedzi.

Ryż. 127. Podwójna warstwa elektryczna
W ogniwach galwanicznych elektroda, która ulega zniszczeniu podczas pracy ogniwa, wysyłając jony do roztworu, nazywana jest anodą, a elektroda, na której wyładowywane są jony dodatnie, nazywana jest katodą.
Ogniwo galwaniczne można zbudować z dowolnych dwóch metali zanurzonych w roztworach ich soli. W tym przypadku absolutnie nie jest konieczne, aby jeden metal był naładowany „ujemnie, a drugi dodatnio. Jedynym warunkiem przepływu elektronów z jednego naładowanego ciała do drugiego jest istnienie między nimi różnicy potencjałów. Ale te ostatnie muszą powstać, cokolwiek by nie było Przyjęli to, ponieważ zdolność odszczepiania elektronów i przechodzenia w jony jest różna dla wszystkich metali. Jeśli na przykład ogniwo galwaniczne jest wykonane z cynku i żelaza zanurzone w normalnych roztworach ich soli, to chociaż oba metale są naładowane ujemnie w roztworach, mimo wszystko pojawi się między nimi pewna różnica potencjałów. Kiedy metale są połączone przewodem, elektrony przepłyną z cynku, jako bardziej aktywnego metalu, do żelaza; rozpuści się i - wyróżni się z rozwiązania. Reakcja zachodząca w elemencie wyrażona jest równaniem
Zn + Fe = Fe + Zn
Różnica potencjałów występująca między metalem a roztworem jego soli nazywana jest potencjałem elektrodowym metalu i może służyć jako miara jego zdolności do oddawania elektronów lub, tym samym, miara jego aktywności chemicznej w reakcjach w rozwiązania. Dlatego mierząc potencjały wszystkich metali przy tych samych stężeniach ich jonów, mogliśmy ilościowo scharakteryzować aktywność metali.
Niestety bezpośredni pomiar tych wielkości jest bardzo trudny i nie daje dokładnych wyników. Wynika to już z faktu, że nie można na przykład podłączyć woltomierza do roztworu bez zanurzenia w roztworze metalowego przewodnika. Ale wtedy istnieje różnica potencjałów między przewodnikiem a roztworem, tak że napięcie wskazywane przez woltomierz będzie zależeć od dwóch różnic potencjałów: różnicy potencjałów między metalem, który nas interesuje a roztworem jego soli, oraz różnicy potencjałów między metalowym przewodnikiem a tym samym rozwiązaniem.
Dużo łatwiej jest zmierzyć różnicę potencjałów (różnicę napięcia elektronów) między dwoma różnymi metalowymi elektrodami zanurzonymi w roztworach odpowiednich soli, czyli dowiedzieć się, o ile potencjał jednego metalu jest większy lub mniejszy niż potencjał innego metalu . Jeśli zmierzymy w ten sposób względne potencjały wszystkich metali, porównując ich potencjały z potencjałem któregokolwiek z nich, to uzyskane liczby będą charakteryzować aktywność metali tak dokładnie, jak bezwzględne wartości ich potencjałów.
Jako standardową elektrodę, z potencjałem, którego porównuje się potencjały innych metali, przyjmuje się tzw. normalną elektrodę wodorową. Ten ostatni składa się z platynowej płytki pokrytej luźną warstwą platyny i zanurzonej w dwunormalnym roztworze kwasu siarkowego. Pod ciągłym ciśnieniem przez roztwór w prądzie 1 atm czysty wodór, który wchodząc w kontakt z platyną jest przez nią wchłaniany w dość dużej ilości. Płyta platynowa nasycona wodorem zachowuje się tak, jakby była wykonana z wodoru. W kontakcie z roztworem kwasu siarkowego powstaje pewna różnica potencjałów (potencjału elektrody wodorowej), umownie przyjmowana jako zero przy pomiarze potencjałów względnych.
Różnica potencjałów między metalem zanurzonym w roztworze jego soli zawierającym 1 gram jonu metalu na litr a normalną elektrodą wodorową nazywana jest normalnym potencjałem metalu.
Aby zmierzyć normalne potencjały, instrumenty podobne do pokazanych na ryc. 128. W istocie takie urządzenie jest ogniwem galwanicznym, którego jedną z elektrod jest badany metal, a drugą jest elektroda wodorowa. Ponieważ potencjał elektrody wodorowej przyjmuje się jako zero, to mierząc różnicę potencjałów na biegunach takiego elementu lub jego siłę elektromotoryczną, znajdujemy bezpośrednio normalny potencjał metalu.
W tabeli. 27 pokazuje normalne potencjały najważniejszych metali. Są one brane ze znakiem minus, gdy potencjał metalu jest poniżej potencjału elektrody wodorowej i ze znakiem plus, gdy potencjał metalu jest powyżej niego.
Jeśli ułożymy metale, w tym i , zgodnie z malejącą wartością napięcia ich elektrod, czyli zgodnie z malejącym ujemnym potencjałem normalnym (i rosnącym dodatnim), otrzymujemy ten sam szereg napięć.
Tabela 27
Potencjały normalne metali
| Metal | I on | Potencjał w woltach | Metal | I on | Potencjał w woltach |
| Do | Do | - 2,92 | Ni | Ni | - 0,23 |
| Sa | Sa | - 2,84 | sn | sn | - 0,14 |
| Na | Na | - 2,713 | Pb | Pb | - 0,126 |
| mg | mg | - 2,38 | n 2 | H | 0,000 |
| Glin | Glin | - 1,66 | Cu | Cu | + 0,34 |
| Mn | Mn | - 1,05 | hg | Hg 2 | + 0,798 |
| Zn | Zn | - 0,763 | Ag | Ag | + 0,799 |
| Fe | Fe | - 0,44 | Au | Au | + 1,42 |
Znając normalne potencjały metali, łatwo jest określić siłę elektromotoryczną dowolnego pierwiastka składającego się z dwóch metali zanurzonych w roztworach ich soli. Aby to zrobić, wystarczy znaleźć różnicę w normalnych potencjałach pobranych metali.
Aby siła elektromotoryczna miała wartość dodatnią, zawsze odejmowana jest mniejsza siła od większego potencjału. Na przykład siła elektromotoryczna elementu miedziano-cynkowego:
mi. s.s. = 0,34 - (-0,763) = 1,103
Oczywiste jest, że będzie miał taką wartość, jeśli stężenia jonów Zn i Cu w odpowiednich roztworach będą równe 1 gramom na 1 litr. Dla innych stężeń potencjały metali, a co za tym idzie siły elektromotoryczne, można obliczyć ze wzoru wyprowadzonego przez Nernsta:
Jeżeli z całego szeregu standardowych potencjałów elektrodowych wyodrębnimy tylko te procesy elektrodowe, które odpowiadają ogólnemu równaniu
wtedy otrzymujemy serię naprężeń metali. Oprócz metali w tej serii zawsze znajduje się wodór, co pozwala zobaczyć, które metale są w stanie wypierać wodór z wodnych roztworów kwasów.
Tabela 19
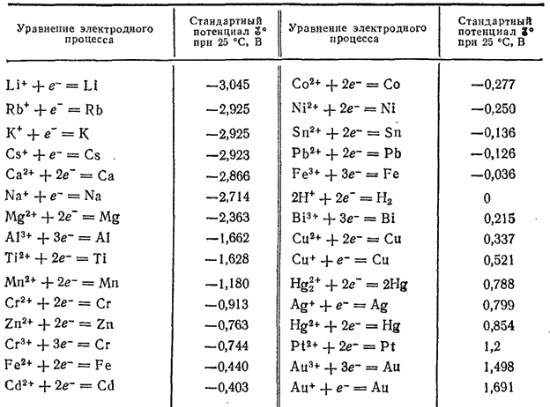
Szereg naprężeń dla najważniejszych metali podano w tabeli. 19. Pozycja metalu w szeregu napięć charakteryzuje jego zdolność do oddziaływań redoks w roztworach wodnych w standardowych warunkach. Jony metali są środkami utleniającymi, a metale w postaci prostych substancji są środkami redukującymi. Jednocześnie im dalej metal znajduje się w szeregu napięć, tym silniejszy środek utleniający w roztworze wodnym to jego jony i odwrotnie, im bliżej znajduje się metal od początku szeregu, tym silniejsza jest redukcja właściwości wykazuje prosta substancja - metal.
Potencjał procesu elektrody
![]()
w neutralnym medium jest to B (patrz strona 273). Aktywne metale na początku szeregu, o potencjale znacznie bardziej ujemnym niż -0,41 V, wypierają wodór z wody. Magnez wypiera jedynie wodór z gorącej wody. Metale znajdujące się pomiędzy magnezem a kadmem zwykle nie wypierają wodoru z wody. Na powierzchni tych metali tworzą się warstwy tlenków, które mają działanie ochronne.
Metale znajdujące się pomiędzy magnezem a wodorem wypierają wodór z kwaśnych roztworów. Jednocześnie na powierzchni niektórych metali tworzą się również folie ochronne, które hamują reakcję. Tak więc film tlenkowy na aluminium sprawia, że metal ten jest odporny nie tylko na wodę, ale także na roztwory niektórych kwasów. Ołów nie rozpuszcza się w kwasie siarkowym w stężeniu poniżej , ponieważ sól powstająca podczas oddziaływania ołowiu z kwasem siarkowym jest nierozpuszczalna i tworzy warstwę ochronną na powierzchni metalu. Zjawisko głębokiego hamowania utleniania metalu, spowodowane obecnością ochronnych warstw tlenku lub soli na jego powierzchni, nazywamy pasywnością, a stan metalu w tym przypadku stanem pasywnym.
Metale są w stanie wypierać się nawzajem z roztworów soli. Kierunek reakcji jest w tym przypadku określony przez ich wzajemne położenie w szeregu napięć. Rozpatrując konkretne przypadki takich reakcji, należy pamiętać, że metale aktywne wypierają wodór nie tylko z wody, ale także z dowolnego roztworu wodnego. Dlatego wzajemne wypieranie metali z roztworów ich soli występuje praktycznie tylko w przypadku metali znajdujących się w rzędzie za magnezem.
Wypieranie metali z ich związków przez inne metale po raz pierwszy szczegółowo zbadał Beketov. W wyniku swojej pracy ułożył metale zgodnie z ich aktywnością chemiczną w serię przemieszczeń, która jest prototypem serii naprężeń metali.
Wzajemne położenie niektórych metali w szeregu napięć iw układzie okresowym na pierwszy rzut oka nie odpowiada sobie. Na przykład, zgodnie z pozycją w układzie okresowym, reaktywność potasu musi być większa niż sodu, a sód musi być większa niż litu. W szeregu napięć lit jest najbardziej aktywny, a potas zajmuje środkową pozycję między litem a sodem. Cynk i miedź, w zależności od ich położenia w układzie okresowym, powinny mieć w przybliżeniu równą aktywność chemiczną, ale w szeregu napięć cynk znajduje się znacznie wcześniej niż miedź. Powód tego rodzaju niespójności jest następujący.
Porównując metale zajmujące określoną pozycję w układzie okresowym, miarą ich aktywności chemicznej – zdolnością redukcyjną – jest wartość energii jonizacji wolnych atomów. Rzeczywiście, podczas przejścia, na przykład od góry do dołu wzdłuż głównej podgrupy grupy I układu okresowego, energia jonizacji atomów maleje, co wiąże się ze wzrostem ich promieni (tj. Z dużą odległością od zewnętrznego elektronów z jądra) oraz wraz ze wzrostem ekranowania dodatniego ładunku jądra przez pośrednie warstwy elektronowe (patrz § 31). Dlatego atomy potasu wykazują większą aktywność chemiczną - mają silniejsze właściwości redukujące - niż atomy sodu, a atomy sodu są bardziej aktywne niż atomy litu.
Porównując metale w szeregu napięć, miarą aktywności chemicznej jest praca polegająca na przekształceniu metalu w stanie stałym w uwodnione jony w roztworze wodnym. Pracę tę można przedstawić jako sumę trzech terminów: energia atomizacji - przemiana kryształu metalu w izolowane atomy, energia jonizacji wolnych atomów metali oraz energia hydratacji powstałych jonów. Energia atomizacji charakteryzuje siłę sieci krystalicznej danego metalu. Energia jonizacji atomów - oderwania od nich elektronów walencyjnych - jest bezpośrednio determinowana przez położenie metalu w układzie okresowym. Energia uwalniana podczas hydratacji zależy od struktury elektronowej jonu, jego ładunku i promienia.
Jony litu i potasu, mające ten sam ładunek, ale różne promienie, będą tworzyć wokół siebie nierówne pola elektryczne. Pole generowane w pobliżu małych jonów litu będzie silniejsze niż pole w pobliżu dużych jonów potasu. Z tego jasno wynika, że jony litu uwodnią się z uwolnieniem większej ilości energii niż jony potasu.
Tak więc w trakcie rozważanej transformacji energia jest zużywana na atomizację i jonizację, a energia jest uwalniana podczas uwodnienia. Im mniejsze całkowite zużycie energii, tym łatwiejszy będzie cały proces i im bliżej początku szeregu napięć będzie się znajdował dany metal. Ale z trzech składników całkowitego bilansu energii tylko jeden - energia jonizacji - jest bezpośrednio określony przez położenie metalu w układzie okresowym. W konsekwencji nie ma powodu, aby oczekiwać, że wzajemne położenie pewnych metali w szeregu napięć zawsze będzie odpowiadało ich położeniu w układzie okresowym. Tak więc w przypadku litu całkowite zużycie energii jest mniejsze niż w przypadku potasu, zgodnie z którym lit znajduje się w szeregu napięć przed potasem.
W przypadku miedzi i cynku nakłady energii na jonizację wolnych atomów i jej pozyskiwanie podczas hydratacji jonów są zbliżone. Ale metaliczna miedź tworzy silniejszą sieć krystaliczną niż cynk, co widać po porównaniu temperatur topnienia tych metali: cynk topi się w , a miedź tylko w . W związku z tym energia zużywana na atomizację tych metali jest znacząco różna, w wyniku czego całkowite koszty energii dla całego procesu w przypadku miedzi są znacznie większe niż w przypadku cynku, co tłumaczy względne położenie tych metali. metale w szeregu napięciowym.
Podczas przechodzenia z wody do rozpuszczalników niewodnych wzajemne położenie metali w szeregu napięć może się zmieniać. Powodem tego jest fakt, że energia solwatacji jonów różnych metali zmienia się w różny sposób podczas przechodzenia z jednego rozpuszczalnika do drugiego.
W szczególności jon miedzi jest bardzo energicznie solwatowany w niektórych rozpuszczalnikach organicznych; prowadzi to do tego, że w takich rozpuszczalnikach miedź znajduje się w szeregu napięć do wodoru i wypiera go z roztworów kwasowych.
Tak więc, w przeciwieństwie do układu okresowego pierwiastków, szereg naprężeń w metalach nie jest odzwierciedleniem ogólnej prawidłowości, na podstawie której można podać wszechstronną Charakterystykę właściwości chemicznych metali. Szereg napięć Charakteryzuje tylko zdolność redox układu elektrochemicznego „metal - jon metalu” w ściśle określonych warunkach: podane w nim wartości dotyczą roztworu wodnego, temperatury i stężenia jednostkowego (aktywności) metalu jony.